









Related Post




How to Minimize Errors in Cross-Linking Mass Spectrometry
2025-09-23Cross-Linking Mass Spectrometry (XL-MS) has powered landmark international studies – from integrative mapping of the human nuclear pore complex to proteome-wide, in-cell analyses using MS-cleavable linkers like DSSO, and rapid SARS-CoV-2 protein interaction work. Applications span structural biology and integrative modeling (restraints for cryo-EM/NMR), native interactomics, quantitative conformational dynamics (QCLMS), chromatin/RNP architecture, epitope and interface mapping for biologics, and mechanism-of-action studies in virology and drug discovery. XL-MS complements classical methods by capturing distance constraints in solution and even in situ, enabling systems-level structural insight.
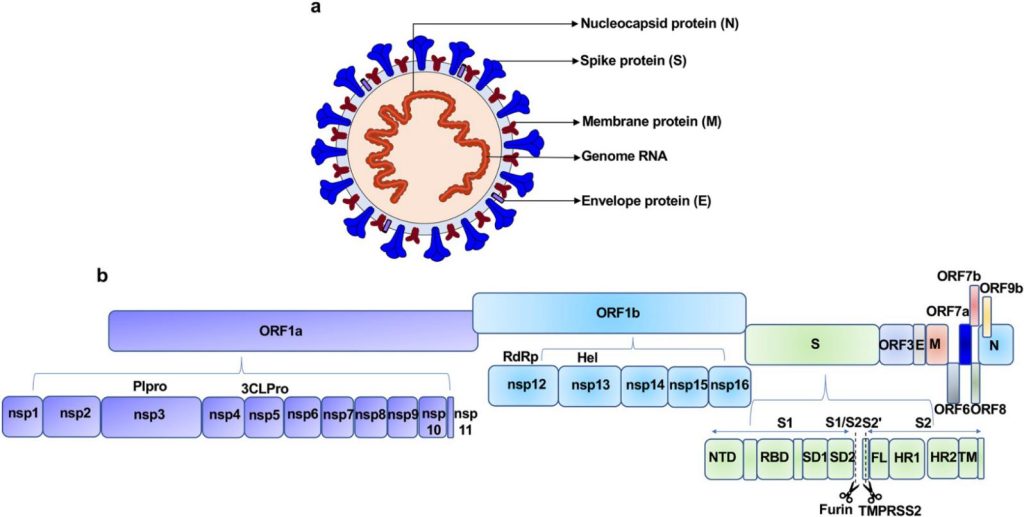
(Molecular mechanism of interaction between SARS-CoV-2
And host cells and interventional therapy | Signal Transduction and Targeted Therapy)
Cross-Linking Mass Spectrometry (XL-MS) is a powerful way to chart protein-protein interactions, yet many labs still grapple with false positives that blur real biology and slow decisions. At Longlight Technology, we tackle the problem at its source – tight chemistry, repeatable prep, and transparent analysis – so you can trust the maps you build.
1) Why False Positives Happen – and How They Creep In
False positives rarely begin at the software stage. They start at the bench. Over-cross-linking pushes proteins into non-physiological pairings. Under-quenching leaves reactive groups that keep linking while you handle the sample. Enzyme digestion that is rushed or inconsistent creates peptide species that masquerade as real cross-links. Add complex matrices and the search space balloons, making borderline spectra look convincing.
The day-to-day reality adds more risk. A buffer prepared slightly off. A quench that runs 5 minutes long. A different enzyme lot. None of these missteps seem dramatic alone, but together they nudge Cross-Linking Mass Spectrometry toward error. The result is a dataset that “looks” rich yet demands hours of triage to find what truly matters.
Despite these pitfalls, XL-MS has unique strengths you don’t want to blunt. Chemical cross-linking coupled with mass spectrometry stabilizes complexes before analysis, preserves short-lived or weak interactions, and provides spatial clues about protein arrangements and connectivity. It supports structural proteomics, can be performed in cells, and does not require special labels. The goal is not to slow the method – it’s to remove noise without losing speed.
✅ Common pain points we see
• Non-specific cross-links formed during sample prep
• Low-abundance cross-links buried by abundant peptides
• Incomplete or uneven digestion that spawns artifacts
• Wide searches paired with lenient filters in data analysis
These issues are solvable with a system view rather than a one-step fix.
2) Longlight‘s System Approach to Reducing False Positives
Our perspective is simple: treat Cross-Linking Mass Spectrometry as a connected chain – chemistry, preparation, detection, and interpretation. We design the cross-linking plan around your biological question and sample type, then keep conditions tight, documented, and repeatable.
✅ Enrichment that Raises Signal Above Noise
A cross-linked peptide is rare by definition. We emphasize selective enrichment after digestion to raise its proportion before the instrument ever sees the sample. Cleaner inputs reduce ambiguous identifications and lower your effective false discovery rate. The effect is practical: better confidence with fewer manual rescues downstream.
✅ Five Habits that Compound into Cleaner Data
• Start with a question-driven cross-linking plan, not a one-size protocol
• Control linking, temperature, and quench window with the same discipline every run
• Standardize digestion and cleanup to limit peptide look-alikes
• Use targeted enrichment to increase cross-linked peptide yield before MS
• Set and document analysis thresholds tied to study goals, then stick to them
This discipline preserves the win conditions of XL-MS: high throughput, intracellular applicability, and no special labeling. When a structure call matters, we recommend pairing Cross-Linking Mass Spectrometry with orthogonal methods such as cryo-EM or X-ray crystallography. The combination narrows interpretation risk from any single technology and helps build models your team can defend.
What “Practical“ Looks like in Your Lab
We don’t ask you to change your science – only to lock in the variables that matter. You get a timestamped plan, reagent checks, and a short list of control points that techs can actually follow. The output is not only cleaner data; it’s a process you can repeat next quarter with new samples and the same confidence.
CTA: Want fewer false positives and sharper interaction maps? Talk to Longlight Technology about a question-driven XL-MS plan that fits your samples and timelines.
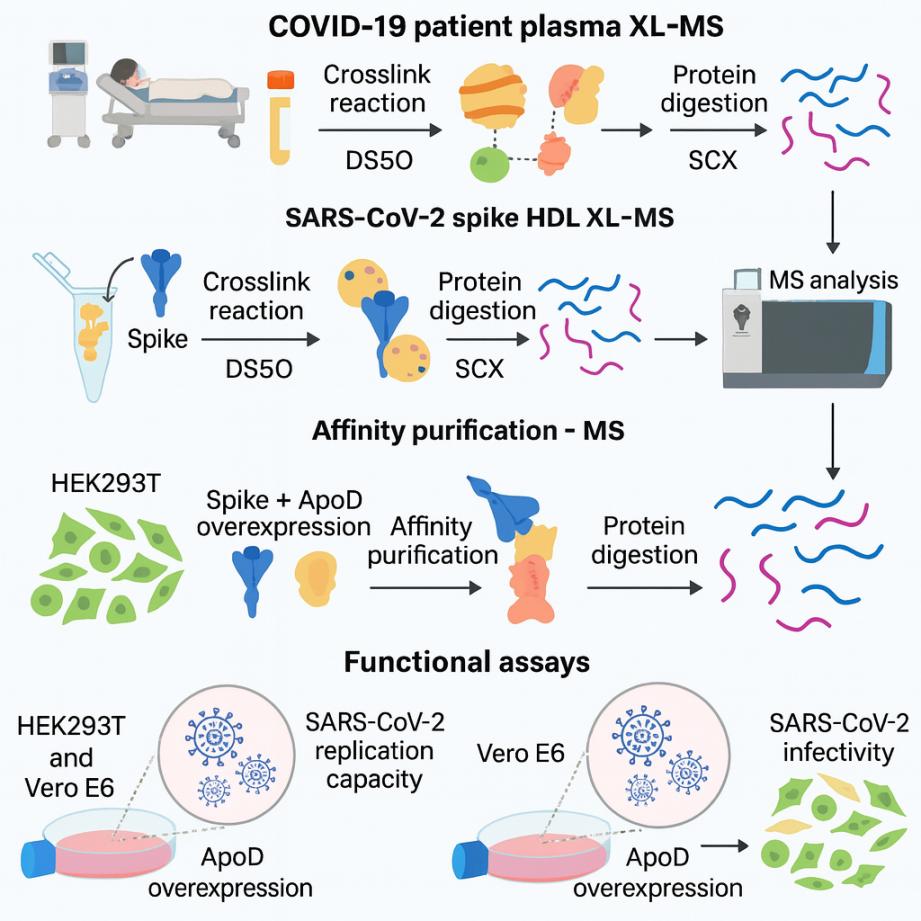
(Cross-Linking Mass Spectrometry Uncovers Interactions Between High-Density Lipoproteins
and the SARS-CoV-2 Spike Glycoprotein – ScienceDirect)
3) From Sample to Report: An End-to-End XL-MS Service You Can Audit
You can ship pre-cross-linked material, or we’ll help design the cross-linking strategy and then receive the samples. From there, our team runs enzyme digestion, selective enrichment, Cross-Linking Mass Spectrometry detection, and structured data analysis. The final report shows interaction networks and cross-link sites with clear methods, so your team can repeat, scale, or integrate with other studies.
What You Receive, Step by Step
Planning → cross-linking strategy → digestion → enrichment → detection → interpretation. Each step includes checkpoints and notes. If something drifts, we can see where and why. That traceability reduces ambiguity and keeps your discovery pace steady without constant firefighting.
Our XL-MS offering sits inside a broader product and service ecosystem designed to keep your lab moving forward. We support modern genomics and proteomics with advanced instruments, reliable reagents, and fit-for-purpose consumables. That continuity matters: the fewer hand-offs between vendors, the easier it is to preserve quality across projects.
Beyond Cross-Linking Mass Spectrometry, we enable complementary applications that many teams run in parallel. For protein-chromatin questions, we support ChIP-seq, combining chromatin immunoprecipitation with next-generation sequencing to pinpoint genomic sites bound by specific transcription factors or histones. For day-to-day workflows, we provide consumables and kits – precast agarose gels, nucleic acid scavengers, Qubit tubes, nucleic acid extraction kits, and library preparation kits – so you can maintain consistent standards from sample intake to data delivery.
Why Teams Choose Longlight Technology
• A single partner for chemistry, workflow design, and XL-MS analysis
• Clear, auditable methods that scale from pilot to production
• Instruments, reagents, and consumables that support genomics and proteomics under one roof
The payoff is not abstract. Fewer false positives mean faster decisions, more credible figures, and less time spent arguing with your own data. Your scientists can focus on biology, not triage. Your stakeholders see progress, not caveats. And your models carry forward into the next program with fewer edits and stronger evidence.
CTA: Ready to tighten your XL-MS pipeline and cut noise at the source? Contact Longlight Technology to schedule a brief consult. We’ll outline a cross-linked peptide enrichment workflow that protects specificity, speeds analysis, and makes your Cross-Linking Mass Spectrometry data easier to trust – run after run.