









Related Post




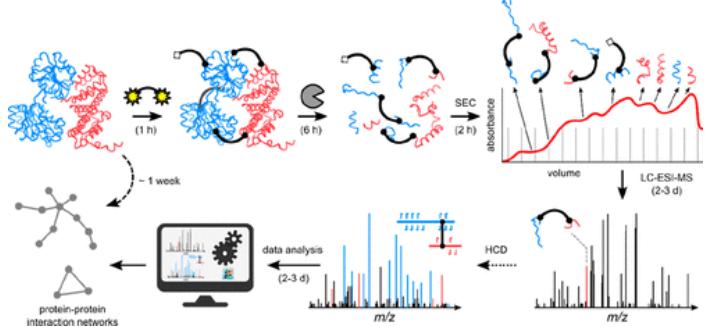
Cross-Linking Mass Spectrometry: FAQs & Proven Solutions
2026-01-15Cross-Linking Mass Spectrometry has rapidly matured from a specialist technique into a cornerstone of structural proteomics and interactome mapping. Early innovators including Ruedi Aebersold, Juri Rappsilber, Andrea Sinz, and collaborators showed how XL-MS can validate cryo-EM models, resolve the organization of large macromolecular machines, and expose fleeting protein – protein contacts that rarely survive purification. Building on this momentum, our integrated ecosystem of services, instruments, and consumables delivers reproducible, high-throughput workflows for Cross-Linking Mass Spectrometry – from sample planning to data interpretation – designed to meet the demands of modern discovery.Cross-Linking Mass Spectrometry (XL-MS) has evolved into a core tool for structural proteomics and interactome analysis. By capturing spatial restraints between residues, XL-MS complements cryo-EM and crystallography and reveals transient protein–protein interactions that often escape purification-based methods.
(Cross-Linking Mass Spectrometry for Investigating Protein Conformations
and Protein-Protein Interactions – A Method for All Seasons)
What Cross-Linking Mass Spectrometry Actually Captures
Cross-Linking Mass Spectrometry (also called chemical cross-linking coupled to mass spectrometry, CL-MS or XL-MS) uses bifunctional reagents to covalently tie together amino acids that sit within a defined spatial window. After cross-linking, targeted enzymatic digestion generates a mixture of linear and cross-linked peptides. Mass spectrometry then detects and identifies these cross-linked species, translating them into distance constraints, interaction partners, and site-specific information that can be fed into structural models. Because XL-MS provides proteomics-derived restraints, it complements cryo-electron microscopy and X-ray crystallography by grounding ambiguous densities and refining interfaces.
Beyond its integrative power, Cross-Linking Mass Spectrometry offers practical advantages: it scales to high throughput, supports intracellular applications, and does not require genetic fusion tags or special chemical labeling. Crucially, the covalent cross-link “freezes” weak or transient contacts, making XL-MS a potent partner to affinity purification or native MS when the goal is to reveal interactions that would otherwise dissipate.
Why Adoption Lags: Persistent Pain Points
Even with clear benefits, several hurdles still impede routine deployment:
-Enrichment and detectability. Cross-linked peptides are typically rare relative to linear peptides. Without thoughtful enrichment or fractionation, identification rates drop and coverage of linkable sites becomes patchy.
-Complex scoring and validation. Composite precursor masses and convoluted fragmentation patterns complicate spectrum interpretation. False discovery rate (FDR) control for cross-linked pairs requires site-aware scoring and carefully tuned thresholds, or spurious links may slip through.
-Chemistry optimization. Cross-linker choice and dose matter. Over-crosslinking can disrupt native architectures; under-crosslinking yields sparse maps. In-cell cross-linking adds matrix complexity that can reduce peptide recovery.
-Instrument method design. Sensitivity must be balanced against fragmentation quality. If you use only HCD, fragment coverage can be uneven; ETD/EThcD needs calibration to ensure the useful ions are recovered for many different peptide pairs.
-Fragmented software and standards. Heterogeneous tools and reporting formats make it difficult to compare results across projects or to merge XL-MS outputs with structural databases for integrative modeling.
Practical FAQs and Proven Solutions for Cross-Linking Mass Spectrometry
- How should I choose a cross–linker for my system?
•Match spacer length to anticipated interaction distances and pick reactive groups aligned with dominant residues (lysine-targeting chemistries are common).
•Pilot titrate cross-linker concentration and reaction duration to prevent over-crosslinking.
•In complex matrices, choose chemistries with well-characterized reactivity and validated quench protocols.
- How can I improve recovery of cross–linked peptides?
• Employ sequential digestion workflows (e.g., Lys-C then trypsin) to broaden peptide boundaries and decrease missed cleavages.
• Use peptide-level enrichment alongside orthogonal fractionation to focus cross-linked species ahead of LC-MS.
- Which LC–MS settings increase identification rates? (No single LC-MS method fits all cross-linked peptides. Method optimization should be considered part of the XL-MS workflow, not a one-time setup.)
•Acquire high-resolution MS/MS and tune collision energies to balance backbone and cross-linker fragmentation.
•Consider mixed fragmentation (HCD supplemented with ETD or EThcD) for richer sequence coverage of cross-linked pairs.
- How do I control false discoveries?
•Apply target – decoy approaches tailored to cross-linked searches and enforce delta score, site-level, and linkage-type filters.
•Validate critical links across biological and technical replicates and cross-check with cryo-EM density or orthogonal biochemical assays.
- Can Cross-Linking Mass Spectrometry capture transient interactions in living cells?
•Yes – cross-linking performed inside cells preserves fleeting interactions. Optimize quenching and lysis for adduct integrity, and use enrichment to manage matrix effects prior to MS.
- How do I integrate XL–MS with cryo–EM or X–ray?
•Provide linker-specific distance bounds and confidence metrics. Use XL-MS restraints to orient domains, confirm interfaces, and flag discordant regions for model refinement.
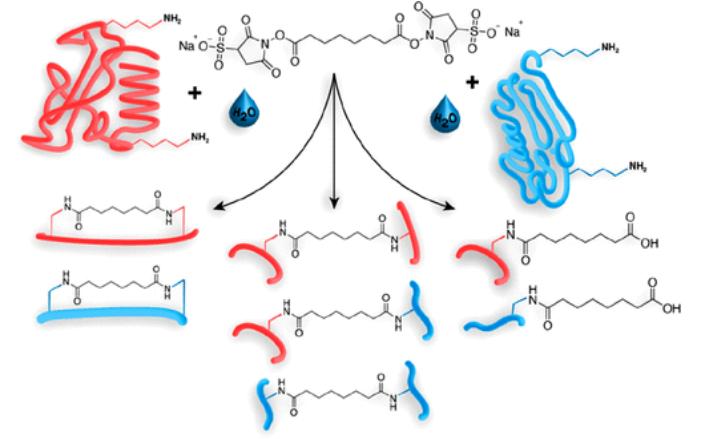
(Reaction of Amine-Reactive NHS Esters, Exemplified for BS3 (Upper Panel).
Reaction Products with Proteins Include Intrapeptide Cross-links (Type 1; Left),
Interpeptide Cross-linksa (Type 2; Middle),
and “Dead-End” Cross-links or “Mono-links” (Type 0; Right))
An Integrated Service and Product Stack That Accelerates XL–MS
We provide an end-to-end XL-MS service, covering experimental design, chemistry optimization,sample preparation, LC-MS acquisition, and data interpretation. This integrated approach minimizes trial-and-error, reduces false discoveries, and shortens the path from pilot experiments to publishable results. Clients may submit pre-cross-linked samples or co-design the workflow with our team from the outset.
- Why Our Genomics Solutions and Laboratory Equipment Strengthen XL–MS?
XL-MS benefits from stable upstream sample prep and robust downstream detection. We provide advanced instruments, high-quality reagents, and consumables that support Cross-Linking Mass Spectrometry and related omics pipelines in academic, clinical, and industrial environments. Driven by efficiency and accuracy, our workflows support high-throughput studies with consistent data quality – ideal conditions for confident interpretation of cross-linked peptide spectra.
- ChIP-Seq and Chromatin Interaction Profiling
When projects interrogate protein – chromatin contacts alongside Cross-Linking Mass Spectrometry, ChIP-seq adds locus-specific genomic context. By mapping transcription factor and histone binding sites genome-wide, ChIP-seq helps connect XL-MS restraints to functional regulatory landscapes, strengthening mechanistic conclusions and guiding hypotheses for follow-up experiments.
- Genomics and Sample Preparation Support
Longlight focuses on molecular biology and molecular diagnostics with a portfolio of NGS-related instruments and reagents, including focused ultrasonication solutions for precise library preparation. These capabilities stabilize upstream sample quality – an often overlooked determinant of success – so that Cross-Linking Mass Spectrometry produces interpretable, reproducible results across batches and projects.
- Consumables and Kits That Reduce Variability
We supply precast agarose gels, nucleic acid scavengers, Qubit tubes, nucleic acid extraction kits, and library preparation kits. Using common consumables and strict standard operating procedures reduces day-to-day variability, helping teams deliver steady XL-MS performance across repeated measurements and multi-omics integrations.
- Streamlined Recommendations to Prevent Common Errors
•Run pilot titrations to dial in reagent concentration and reaction time per matrix.
•Use standardized protease workflows and peptide-level enrichment to increase cross-linked peptide yield.
•Keep LC – MS method versions logged and, when appropriate, favor high-resolution hybrid HCD/ETD or EThcD fragmentation.
•Set strict score, delta-score, and site-level filters, and reproduce priority links in independent technical and biological replicates.
•Combine Cross-Linking Mass Spectrometry with cryo-EM or X-ray and disclose linker-specific distance bounds and confidence metrics for integrative modeling.
From Pilot to Scale: Your Next Step
Planning Cross-Linking Mass Spectrometry for protein interaction mapping, structure validation, or integrative modeling? Team up with our experts to design a dependable, high-throughput workflow optimized for your goals. We will advise on cross-linker selection, organize sample submission, and perform digestion, enrichment, MS acquisition, and data analysis. Expect a concise report with actionable insights. Collaborate with us to cut risk, fix common bottlenecks, and elevate XL-MS from early pilots to dependable, day-to-day discovery.